Bioinformática para descifrar secuencias genéticas: entrevista a Luca Cozzuto




Luca Cozzuto estudió biotecnología en la Universidad de Nápoles. A posteriori se embarcó en un doctorado en medicina molecular que le llevó a trabajar en la Unidad de Bioinformática del CRG como responsable de análisis de datos genómicos. Aquí trabaja desde hace 8 años. La Unidad la forman un equipo de cuatro personas que ofrecen asistencia y apoyo a los científicos para el análisis de datos de sus experimentos o el desarrollo de software y bases de datos para la investigación.
Luca, ¿en qué momento de la investigación los datos de Saca La Lengua pasan por tus manos?
Los datos nos llegan recién salidos del secuenciador: nosotros somos los encargados de analizarlos para descifrar las secuencias genéticas e identificar y cuantificar los géneros y familias de bacterias presentes en las muestras de saliva.
¡Explícanos cómo los trabajas!
El primer paso es organizar los datos. El secuenciador proporciona miles de secuencias de ADN bacteriano, que en lenguaje técnico llamamos “reads”, o sea, lecturas.
Para que os hagáis una idea, para 1.500 muestras de saliva estamos hablando de casi 90.000 reads que corresponden a unos 54.000.000.000 nucleótidos, que representan unos 120 Gigabytes de datos.
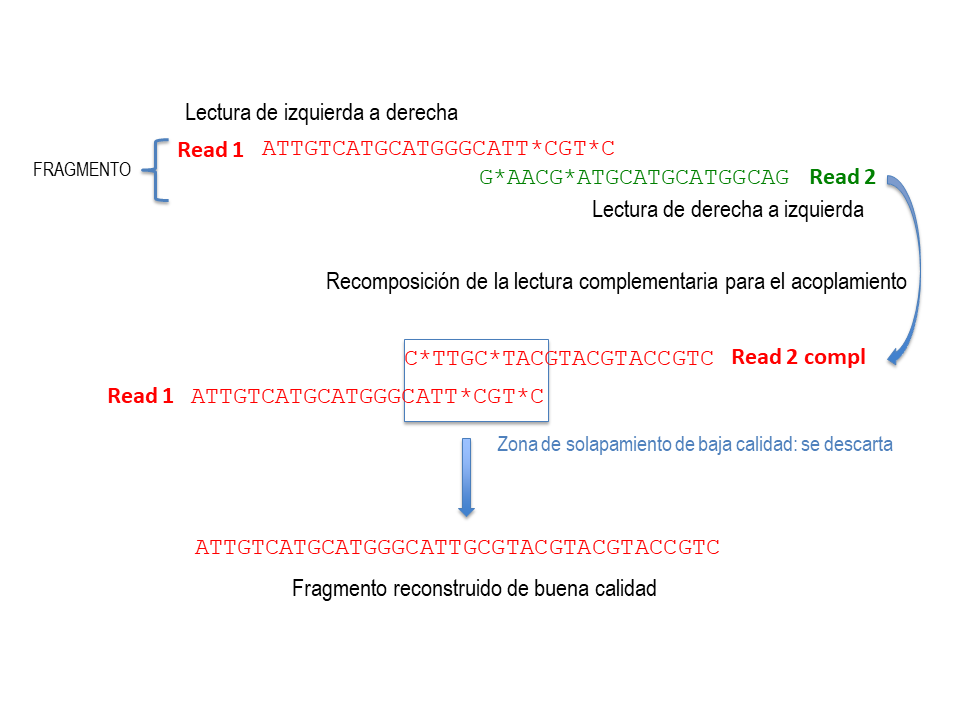
Estas secuencias son el resultado de una “doble” lectura de cada fragmento de ADN, procedimiento estándar para asegurar la buena calidad de los datos.
¿Doble lectura? ¿Para qué se hace?
El secuenciador es capaz de leer el ADN de las muestras, pero empieza con mucha calidad y a medida que avanza se va haciendo menos preciso. Es como si la máquina estuviera leyendo y con el tiempo, se fuera “cansando” y leyera peor.
Para asegurar calidad, lo que hacemos es leer la misma secuencia dos veces, primero en un sentido, y luego en el otro. En los dos casos, solo nos quedaremos con las primeras partes de estas lecturas, las de máxima calidad, y descartaremos la parte de peor calidad, que se solapa una vez unidos los fragmentos. (ver la imagen a la derecha)
En esta primera fase del trabajo, también se descartan las secuencias que pertenecen a porciones conservadas del ADN (o sea que son comunes a todas las bacterias, por lo que no son útiles para distinguir diferencias entre familias o especies), y las “quimeras” (secuencias erróneas, producidas por el método de reacción en cadena de la polimerasa [PCR], que no corresponden a ningún ser vivo).
¿Y después?
Una vez descartados estos secuencias artificiales, y reconstruidos los fragmentos de ADN original a partir de las dos lecturas, viene lo interesante: comparar este conjunto de secuencias con las de una base de datos de ADN ribosómico (que es el que estamos utilizando para este estudio). Este proceso permitirá identificar a qué grupos (géneros o familias) de bacterias corresponden las secuencias obtenidas y deducir la proporción de estos grupos en la saliva de cada persona.
Para este estudio hemos utilizado SILVA, una base de datos de secuencias de ADN ribosómico para los tres dominios de la vida (Bacteria, Archaea y Eukarya), desarrollada y regularmente actualizada por un grupo de investigación alemán.
Seguramente, algunos se preguntarán porque simplemente no se miran las muestras al microscopio…
El problema es que las bacterias, al microscopio, no presentan grandes diferencias. Es decir, por fuera, se parecen todas bastante y no las podríamos distinguir. En cambio, pertenecen a especies, géneros y familias diferentes, y tienen metabolismos y funciones diferentes.
Gracias a la existencia de bases de datos como SILVA, que están a disposición de la comunidad científica, podemos descifrar las especies, géneros o familias de estos microorganismos, analizando solo un pequeño fragmento y no todo su ADN. Aún así, estamos hablando de un trabajo que supone utilizar ordenadores bastante potentes, por ejemplo, con una RAM de 512 GB, 8 CPUs y ¡5 terabytes de disco duro para almacenar los datos!
Los “fragmentos” que analizamos serían el equivalente al código de barras de un producto que compramos en el supermercado. Aquí está la información base para saber que es una bacteria (y no un animal, por ejemplo), y que corresponde a una especie y no a otra.
Este proceso resulta más rápido y más económico que si tuviéramos que leer todo el genoma de todas las bacterias presentes. En el caso de Saca La Lengua por ejemplo, sería como leer más de 600 genomas que tienen una media de 4 millones de bases cada uno.
¿Qué reto ha supuesto para ti el proyecto Saca La Lengua?
Con Saca la Lengua nos enfrentamos por primera vez a datos de microbioma. Poner a punto la técnica, ha llevado su tiempo, así como testar e instalar los programas necesarios para el análisis. Ha sido un reto desde el principio.
A nivel personal, me ha implicado también en otros ámbitos, no puramente científicos, en los que no estaba acostumbrado a moverme. Por ejemplo, hacer cursos de bioinformática para profesores de secundaria, explicar mi trabajo delante de público no especializado y sin soporte alguno en la Fiesta de la Ciencia de Barcelona, o participar en la cocreación de un juego basado en los resultados del proyecto. Me ha gustado ver cómo este proyecto ha impactado en la sociedad y he disfrutado saliendo de mi “zona de confort”.
Para acabar, dinos qué te gusta más de tu profesión. ¿Aconsejarías a los jóvenes dedicarse a la bioinformática?
Lo que más me gusta de mi profesión es que plantea retos cada día. No hay una sola forma de solucionar un problema: en bioinformática tienes que encontrar la mejor manera de afrontar el reto con las herramientas existentes o desarrollar nuevas herramientas para contestar a preguntas biológicas concretas, que plantea la investigación.
La bioinformática está en auge y cada día requieres de más profesionales, porque cada día se producen cantidades masivas de datos (big data) y se necesitan superordenadores y programas específicos para analizarlos. Se trata de un sector en rápida evolución, y con muchas posibilidades. Animaría a cualquier persona que tenga curiosidad y le apasionen los retos a probarlo. ¡Seguro que se engancha!